with open("sets.txt") as f:
print(f.read())ChiD1T
ChiD1D
ChiD2T
ChiD2D
ChiW1T
ChiW1D
ChiW2T
ChiW2D
LimnD1T
LimnD1D
LimnD2T
LimnD2D
LimnW1T
LimnW1DWelcome to this reproducible project, where we follow the MerenLab tutorial guide to recover, refine, and characterize metagenome-assembled genomes (MAGs) from two Antarctic lake catchments (Byers Peninsula).
In this step, we performed quality filtering of raw reads from Limnopolar and Chica soil samples using illumina-utils.
The input data are the raw paired-end reads (*.fastq.gz) stored in data/raw/.
We will be working with 16 metagenomic sets, 8 for each basin. There are 48 samples in total, each one has the corresponding prefix from ‘sets.txt’, followed by a letter indicating the plot they come from (A, B or C).
The contents of the sets file should look like this:
Quick note: adjust the paths as you wish.
with open("sets.txt") as f:
print(f.read())ChiD1T
ChiD1D
ChiD2T
ChiD2D
ChiW1T
ChiW1D
ChiW2T
ChiW2D
LimnD1T
LimnD1D
LimnD2T
LimnD2D
LimnW1T
LimnW1DA samples.txt file will be also required. This file contains the sample name, followed by the compressed reads file:
with open("samples.txt") as f:
print(f.read())Sample r1 r2
ChiD1T /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1T_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1T_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1T_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1T_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1TB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1TC_2.fastq.gz
ChiD1D /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1D_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1D_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1D_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1D_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1DB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD1DC_2.fastq.gz
ChiD2T /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2T_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2T_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2T_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2T_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2TB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2TC_2.fastq.gz
ChiD2D /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2D_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2D_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2D_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2D_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2DB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiD2DC_2.fastq.gz
ChiW1T /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1T_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1T_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1T_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1T_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1TB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1TC_2.fastq.gz
ChiW1D /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1D_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1D_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1D_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1D_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1DB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW1DC_2.fastq.gz
ChiW2T /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2T_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2T_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2T_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2T_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2TB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2TC_2.fastq.gz
ChiW2D /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2D_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2D_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2D_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2D_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2DB_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/ChiW2DC_2.fastq.gz
LimnD1T /home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_B_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD1T_C_2.fastq.gz
LimnD2D /home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_A_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_B_1.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_C_1.fastq.gz /home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_A_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_B_2.fastq.gz,/home/meridian/MERIDIAN_2024/Samples_HN00251955/LimnD2D_C_2.fastq.gzWe will work with the program iu-gen-configs on the samples.txt file:
iu-gen-configs samples.txtThe program iu-filter-quality-minoche performs quality filtering of raw Illumina reads based on the method described by Minoche et al. (2011).
Briefly, it removes reads and base pairs that are likely to contain sequencing errors or low-quality regions. This helps to reduce noise and improve the accuracy of downstream analyses (assembly and binning).
for sample in `awk '{print $1}' samples.txt`
do
if [ "$sample" == "sample" ]; then continue; fi
iu-filter-quality-minoche $sample.ini --ignore-deflines
doneThis step generates files that summarize the quality filtering results for each sample, as well as the FASTQ files for each sample that contain quality-filtered reads:
#with open("ChiD1T-STATS.txt") as f:
# print(f.read())Before we move on with the co-assemblies, we are going to get rid of possible contaminated reads originated from human genome, since human contamination is very frequent. On the other hand, we also want to eliminate the phiX174 genome. It was the first genome to be sequence Sanger, F., Air, G., Barrell, B. et al. Nucleotide sequence of bacteriophage φX174 DNA. Nature 265, 687–695 (1977). Since that, the viral genome is included as an spike by Illumina kits to control quality of the sequencing process and some contaminating reads could be left.
To perform short read alignment we are going to use [Bowtie2(https://bowtie-bio.sourceforge.net/bowtie2/index.shtml)]. There are other popular aligners such as BWA, but Bowtie2 parameters are easy to understand and modify. Specially, as we are not really interested in aligned reads, those that mapped to the human or phiX174 genomes, by using Bowtie2 we have an option to extract not aligned reads easily. We will use the fasta files generated in last step: *-QUALITY_PASSED_R*.fastq.gz
I recommend using conda as your installer and enviroment manager:
conda create -n bowtie2 -c bioconda bowtie2 -ywget https://ftp.ncbi.nlm.nih.gov/genomes/refseq/vertebrate_mammalian/Homo_sapiens/latest_assembly_versions/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_cds_from_genomic.fna.gz
gunzip GCF_000001405.40_GRCh38.p14_cds_from_genomic.fna.gz
bowtie2-build GCF_000001405.40_GRCh38.p14_cds_from_genomic.fna.gz human_cdsfor SET in `cat sets.txt`
do
bowtie2 -x human_cds \
-1 ${SET}*-QUALITY_PASSED_R1.fastq.gz \
-2 ${SET}*-QUALITY_PASSED_R2.fastq.gz \
--un-conc ${SET}_nohuman_R%.fastq \
-S ${SET}_tmp.sam \
-p 28
done# Download from NCBI and decompress
wget https://ftp.ncbi.nlm.nih.gov/genomes/refseq/viral/Sinsheimervirus_phiX174/latest_assembly_versions/GCF_000819615.1_ViralProj14015/GCF_000819615.1_ViralProj14015_genomic.fna.gz
gunzip GCF_000819615.1_ViralProj14015_genomic.fna.gz
# Building Bowtie2 index
bowtie2-build GCF_000819615.1_ViralProj14015_genomic.fna phix
# Aligning human decontaminated reads against PhiX174 index
for SET in `cat sets.txt`
do
bowtie2 -x phix174 \
-1 ${SET}_nohuman_R1.fastq \
-2 ${SET}_nohuman_R2.fastq \
--un-conc ${SET}_nohuman_noPhiX_R%.fastq \
-S /dev/null \
-p 28
done
# Count decontaminated reads
for SET in `cat sets.txt`
do
echo ">>> $SET"
wc -l ${SET}_nohuman_noPhiX_R*.fastq | awk '{print $1/4}'
done
# We simplify file names
for SET in `cat sets.txt`
do
mv ${SET}_nohuman_noPhiX_R1.fastq ${SET}_clean_R1.fastq
mv ${SET}_nohuman_noPhiX_R2.fastq ${SET}_clean_R2.fastq
doneNow we have cleaned and trimmed reads, with no human genome contamination and no phage phix174. Hurray!
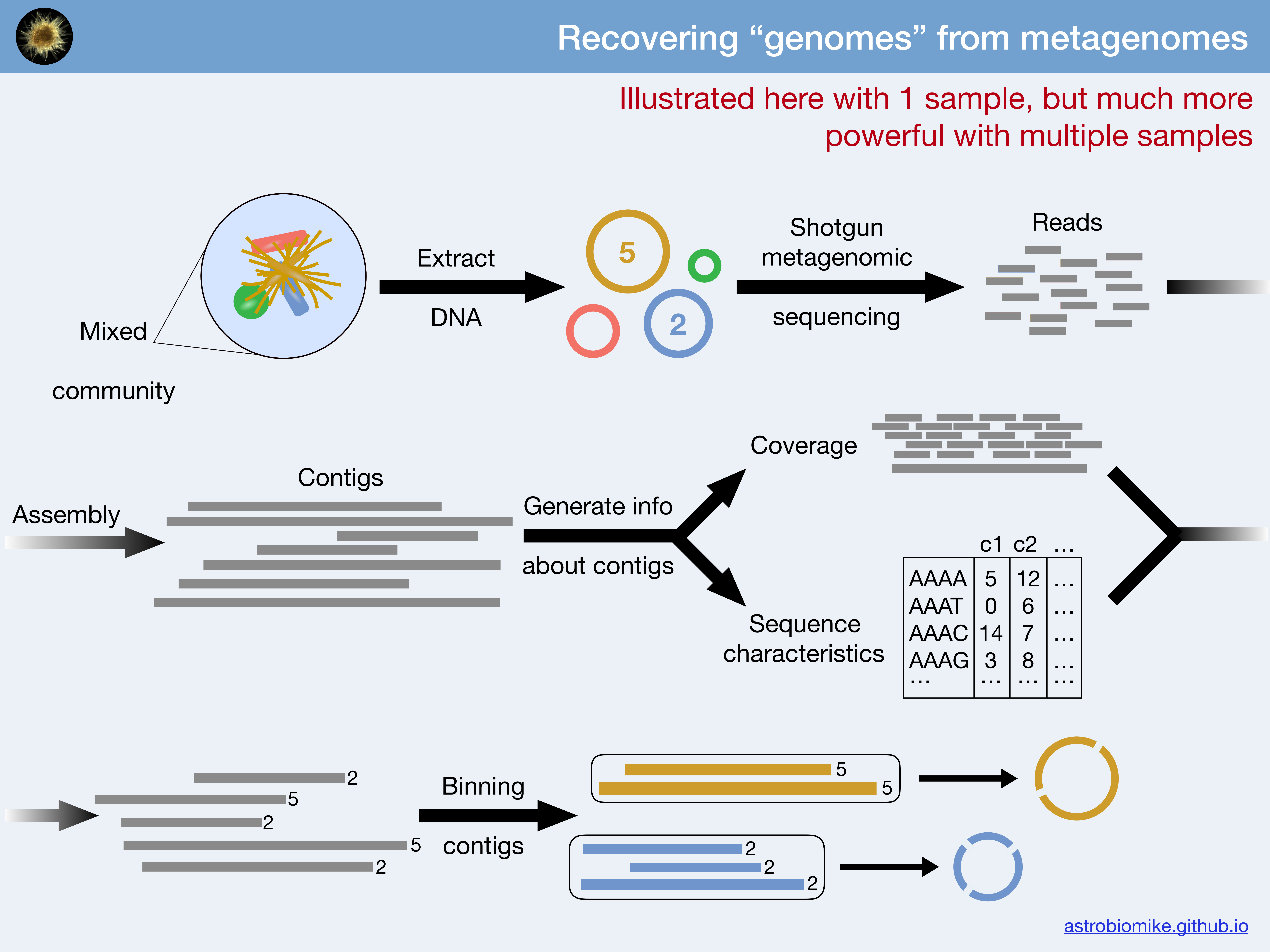
“Co-assembly” is the computational process of assembling DNA sequencing reads from multiple biological samples into a single, combined reference assembly, as opposed to performing an independent assembly for each sample. This approach brings many benefits:
As Happy Belly Bioinformatics points out, co-assembly might seem like a good general choice to follow, but it won’t be ideal in all circumstances. From my personal experience, the golden rule in bioinformatics is that there is no golden rule as for when it would be better to co-assemble multiple samples together over when it would be better to run individual assemblies on each. The outcome can depend on many factors, such as variation among samples, community diversity, the assembler you choose, the biological questions you are addressing, and many more. In some cases, a co-assembly may actually result in a lower-quality assembly than individual assemblies. For this reason, it’s important to evaluate each dataset on its own. If you have the time and resources, testing both approaches and comparing the results is always good practice.
In our case, since samples belong to the same study site but are subjeted to different enviromental conditions (humidity, MO(%), depth), they are more likely to share a portion of their sequence by space. By performing co-assemblies across each 3 biological replicates, the mean coverage of microbial genomes can be increased, which in turn improves the completeness and resolution of the recovered genomes (MAGs). This strategy takes advantage of the overlap between replicates while still capturing condition-specific diversity, providing a more robust genomic representation of the microbial communities.
Since we are going to work with a large dataset, I prefer to use MEGAHIT (Liu et al., 2015) as our assembler, since it can handle large ammounts of information better than SPADES. Syntax is as follows:
for SET in `cat sets.txt`
do
megahit -1 ${SET}_clean_R1.fastq \
-2 ${SET}_clean_R2.fastq \
--min-contig-len 1000 \
-o $SET-RAW.fa \
--num-cpu-threads 28
doneA good practice is to rename contig names to simplify header names of all scafolds. We want to rename headers with the ´set´name they belong to. This will make downstream analyses much easier to follow and understand. Anvio also likes easy names ;). We can use the command anvi-script-reformat-fasta as follows:
for SET in $(cat sets.txt)
do
DIR="$SET-RAW.fa"
FILE="$DIR/$SET-RAW.fa"
if [[ -f "$FILE" ]]; then
anvi-script-reformat-fasta "$FILE" \
--simplify-names \
-o "$DIR/$SET-RAW-FIXED.fa" \
--prefix "$SET"
# Overwrite the temporal file (-FIXED) with original names
mv "$DIR/$SET-RAW-FIXED.fa" "$FILE"
else
echo "$FILE not found"
fi
doneI recommend creating a folder named “assemblies-1000nt” and using symbolic links to all renamed assemblies files. For that, i created a bash script:
You can also move directly the files to a new folder. I just prefer to keep files in their original location.
ASSEMBLY_DIR="/media/meridian/Elements/MERIDIAN_2024/02_assembly"
LINK_DIR="$ASSEMBLY_DIR/assemblies-1000nt"
mkdir -p "$LINK_DIR"
for dir in "$ASSEMBLY_DIR"/*.fa; do
if [ -d "$dir" ]; then
base=$(basename "$dir")
target="$dir/$base"
if [ -f "$target" ]; then
ln -s "$target" "$LINK_DIR/$base"
echo "Link created: $LINK_DIR/$base -> $target"
fi
fi
doneBoth the tetra-nucleotide frequency and differential mean coverage of scaffolds across metagenomes are instrumental to accurate binning of scaffolds into metagenomic bins. To prepare for the binning step, we first removed scaffolds too short to produce a reliable tetra-nucleotide frequency using the program ´anvi-script-reformat-fasta´, and then used the program ´anvi-gen-contigs-database´ with default parameters to profile all scaffolds and generate an anvi’o contigs database that stores the nucleic acid sequence, tetra-nucleotide frequency, GC-content, and the coordinates of genes Prodigal v2.6.3 (Hyatt et al. (2010)) identified:
for SET in `cat sets.txt`; do
anvi-script-reformat-fasta \
.../assemblies-1000nt/$SET-RAW.fa \
--min-len 2500 \
--simplify-names \
-o $SET.fa
anvi-gen-contigs-database \
-f $SET.fa \
-o $SET-CONTIGS.db \
-T 28
doneNow that we have our contigs databases, we can add some basic information about our sequences. Using HMMER to scan for a commonly used set of single-copy genes. Most microbial genomes contain exactly one copy of each single-copy core gene (SGC) in the set of known SGCs for their taxonomic domain. So, the number of HMM hits to a known SGC in a metagenome can serve as an estimate of the number of genomes in that sample (for a given taxonomic domain). Furthermore, this step will help us estimate genome completeness/redundancy in real-time as we work on binning our contigs:
for SET in `cat sets.txt`
do
anvi-run-hmms -c $SET-CONTIGS.db --num-threads 28
doneWe can import taxonomy into anvio with pretty much any software, although anvio has specific parsers to make sense of the output of Kaiju and Centrifuge. We will use centrifuge, assigning taxonomy to the open-reading frames prodigal predicted. First, we will use the program ´anvi-get-dna-sequences-for-gene-calls´ to export nucleic acid sequences corresponding to the gene calls from each anvi’o contigs database. Then we will use Centrifuge and an index of NCBI’s nr database to assess the taxonomy of genes, and finally, the ´program anvi-import-taxonomy´ to import this information back into the anvi’o contigs databases.
for SET in `cat sets.txt`
do
anvi-get-sequences-for-gene-calls -c $SET-CONTIGS.db -o $SET-gene_calls.fa
done
centrifuge -f -x /path/to/nr_index/nr $SET-gene_calls.fa -S $SET-centrifuge_hits.tsv -p 28
doneThen we can import them into our contigs databases:
for SET in `cat sets.txt``
do
anvi-import-taxonomy-for-genes -c $SET-CONTIGS.db -i $SET-centrifuge_hits.tsv -p centrifuge
doneNow is a good time to poblate our contigs databases with functional annotation of the open-reading frames prodigal predicted. My experience has taught me that using a variety of databases is the best practice to ensure that we are not being overly sensitive to the lacking of information. For this reason, we will use NCBI COGs and KEGG for functional annotation. First, we need to set up our COG and KEGG for anvi’o:
# COGs
anvi-setup-ncbi-cogs -T 20 --just-do-it
for SET in `cat sets.txt`
do
anvi-run-ncbi-cogs -c $SET-CONTIGS.db -T 20
done# KEGGs
anvi-setup-kegg-data -T 20 --just-do-it
for SET in `cat sets.txt`
do
anvi-run-kegg-kofams -c $SET-CONTIGS.db -T 20
doneI am a guy who really likes numbers and statistics. Lucky for me, contigs statistics are crucial in genome assembly because they give us important information about the quality and structure of the assembly. Metrics such as the total number of contigs, N50, L50, maximum contig length, and length distribution help assess whether the assembly is sufficiently contiguous to support reliable binning. They also reveal the proportion of short versus long contigs, which is important since binning algorithms rely on sequence composition and coverage signals that are more meaningful in longer contigs. Without this assessment, you risk using assemblies that are too fragmented, introducing noise, or setting inappropriate minimum contig length thresholds, all of which can compromise the quality and interpretability of the recovered genomes (MAGs).
We can obtain contigs statistics by using the program ´anvi-display-contigs-stats´:
anvi-display-contigs-stats 03_contigs_dbs/*.db \
--report-as-text \
--as-markdown \
-o all_contigs_statistics.mdWhich will show something like:
| contigs_db | ChiD1T | ChiW1T | IZBWet | LimnW1T |
|---|---|---|---|---|
| Total Length | 657351660 | 376227450 | 142891251 | 708536870 |
| Num Contigs | 114608 | 72268 | 28763 | 129590 |
| Num Contigs > 100 kb | 114 | 14 | 0 | 94 |
| Num Contigs > 50 kb | 643 | 145 | 36 | 501 |
| Num Contigs > 20 kb | 3247 | 1338 | 459 | 3081 |
| Num Contigs > 10 kb | 10529 | 5588 | 1987 | 10667 |
| Num Contigs > 5 kb | 34805 | 20727 | 7542 | 37217 |
| Num Contigs > 2.5 kb | 114608 | 72268 | 28763 | 129590 |
| Longest Contig | 441616 | 201055 | 95413 | 336007 |
| Shortest Contig | 2500 | 2500 | 2500 | 2500 |
| Num Genes (prodigal) | 724637 | 420527 | 155248 | 791048 |
| L50 | 23973 | 17874 | 7446 | 29016 |
| L75 | 59451 | 40000 | 16307 | 69315 |
| L90 | 90041 | 58085 | 23342 | 102965 |
| N50 | 6200 | 5426 | 5031 | 5742 |
| N75 | 3656 | 3477 | 3353 | 3541 |
| N90 | 2875 | 2826 | 2787 | 2844 |
| Archaea_76 | 5087 | 3351 | 798 | 6226 |
| Bacteria_71 | 10902 | 6992 | 1567 | 13103 |
| Protista_83 | 777 | 490 | 187 | 875 |
| bacteria (Bacteria_71) | 147 | 98 | 22 | 152 |
| eukarya (Protista_83) | 0 | 0 | 0 | 0 |
| archaea (Archaea_76) | 0 | 1 | 0 | 1 |
We can then use pandoc to convert markdown tables to PDF or HTM.
pandoc -V geometry:landscape \
all_contigs_statistics.md
-o OUTPUT_FILE_NAME.pdfThis is just an example. Since we have high number of metagenomic sets, the table will be difficult to fit in a pdf page. HTML seems like the reasonable choice for us.
for SET in `cat sets.txt`
do
bowtie2-build $SET.fa $SET
donefor sample in `awk '{print $1}' samples.txt`
do
if [ "$sample" == "sample" ]; then continue; fi
# determine which metagenomic set the $sample belongs to:
SET=`echo $sample | awk 'BEGIN {FS="_"}{print $1}'`
# do the bowtie mapping to get the SAM file:
bowtie2 --threads 24 \
-x $SET \
-1 $sample_clean_R1.fastq \
-2 $sample_clean_R2.fastq \
--no-unal \
-S $sample.sam
# covert the resulting SAM file to a BAM file:
samtools view -F 4 -bS $sample.sam > $sample-RAW.bam
# sort and index the BAM file:
samtools sort $sample-RAW.bam -o $sample.bam
samtools index $sample.bam
# remove temporary files:
rm $sample.sam $sample-RAW.bamfor sample in `awk '{print $1}' samples.txt`
do
if [ "$sample" == "sample" ]; then continue; fi
# determine which metagenomic set the $sample bleongs to:
SET=`echo $sample | awk 'BEGIN {FS="_"}{print $1}'`
anvi-profile -c $SET-CONTIGS.db \
-i $sample.bam \
--num-threads 16 \
-o $sample
donefor SET in $(cat sets.txt); do
path/to/jgi_summarize_bam_contig_depths \
--outputDepth ${SET}_metabat.txt \
/media/meridian/Elements/MERIDIAN_2024/04_read_recruitment/${SET}.bam
done